BrainGnomes Quickstart
Michael Hallquist
31 Aug 2025
Source:vignettes/braingnomes_quickstart.Rmd
braingnomes_quickstart.RmdIntroduction
BrainGnomes is an R package that streamlines the preprocessing and analysis of fMRI data on high-performance computing (HPC) clusters. It serves as a wrapper around common fMRI processing tools such as HeuDiConv (for DICOM-to-BIDS conversion), BIDS Validator, MRIQC (quality control), fMRIPrep (preprocessing pipeline), and ICA-AROMA (automatic removal of motion artifacts), orchestrating their execution in a coherent pipeline. The package also provides additional processing steps for preparing fMRI data for analysis, such as spatial smoothing, temporal filtering, and confound regression. BrainGnomes uses containerized software (Singularity images) to ensure reproducible environments for imaging tools and it manages job submission to HPC schedulers (SLURM or TORQUE). In this tutorial, we will walk through the full workflow of setting up and running an fMRI preprocessing study using BrainGnomes. We will cover the three key functions:
- setup_project(): Initialize a new study configuration (either from scratch or from an existing YAML configuration file)
- edit_project(): Interactively review and modify the study configuration by sections
- run_project(): Execute the preprocessing pipeline steps on your dataset, utilizing the HPC environment
Along the way, we assume you have the required Singularity container images for each tool and a working HPC environment. Additional information about setting up the compute environment is provided in BrainGnomes Singularity container setup. This vignette provides example R code snippets, explains expected inputs/outputs, and offers troubleshooting tips for common issues. By the end of demo, a new user should understand how to configure a study and launch the BrainGnomes pipeline from start to finish.
This package is designed to run on HPC clusters that use SLURm or TORQUE. It not designed to run on a standard computer.
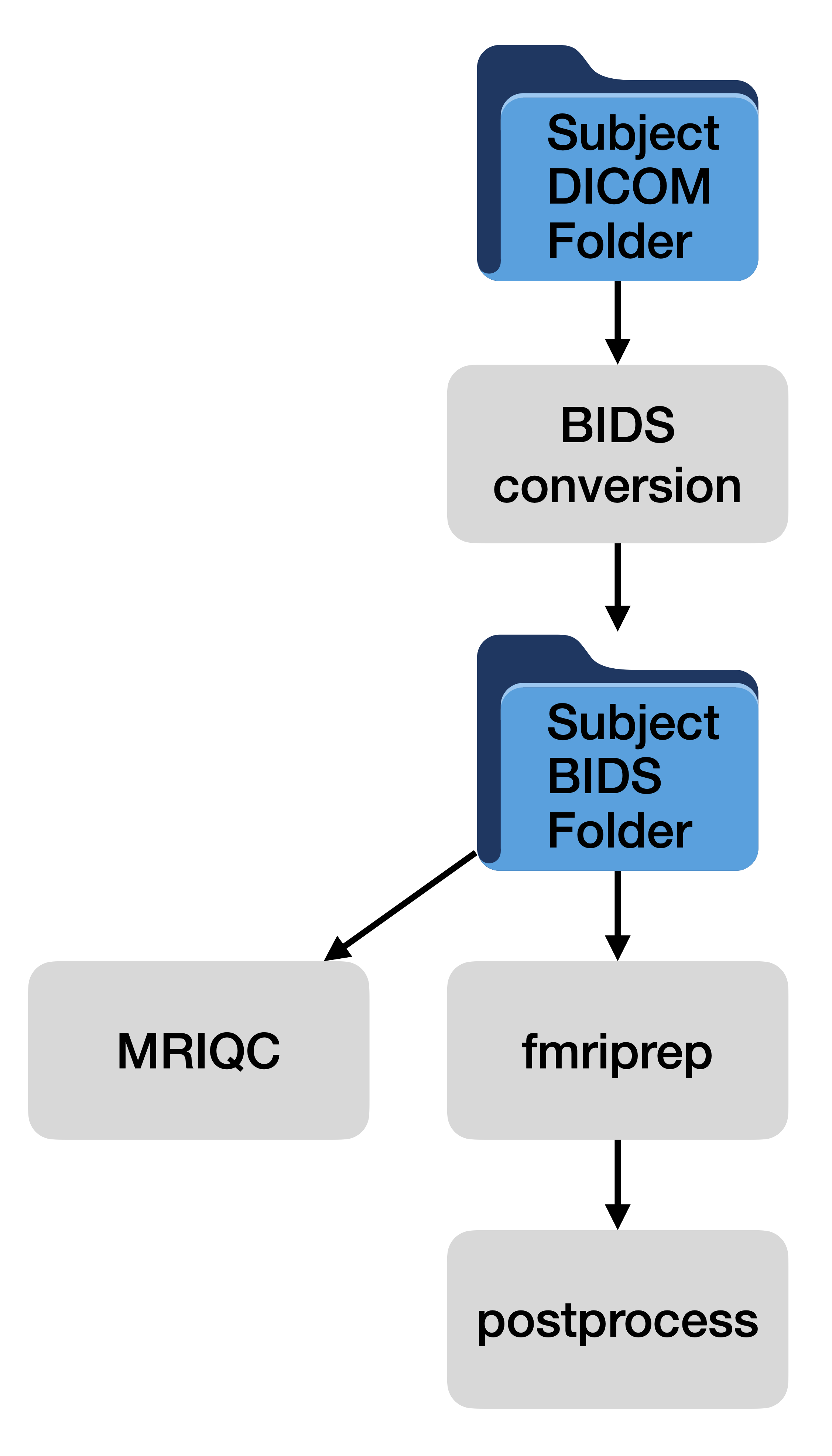
Basic process flow of BrainGnomes
BrainGnomes identifies data for all subjects (and sessions) in your DICOM and BIDS folders, then determines which subjects need to be processed. It processes data by looping over subjects, submitting jobs for each subject and processing step to the HPC scheduler. This ensures that each subject is processed fully (unless a crash occurs).
Running a subset of subject or a subset of processing steps
As detailed below, the run_project function submits fMRI
processing jobs to the scheduler. Although BrainGnomes is broadly
intended to run all processing steps and all subjects, you can also run
a subset of subjects. In the intractive mode of
run_project, you can enter a set of subject IDs to run in
case you don’t want to run all of them.
As detailed below, a BrainGnomes project can be configured to only
run some steps (e.g., fmriprep and postprocessing). In this case, you
will only be asked about these steps in run_project. Even
if you have enabled a step, however, you can choose not to run it in
run_project.
For example, if you only wish to run postprocessing stream “postproc1” on subject 540311, you might respond to the prompts like this:
Enter subject IDs to process, separated by spaces. Press enter to process all subjects.
(Press enter to skip) > 540311
Please select which steps to run:
Run MRIQC?
(yes/no) > n
Run fmriprep?
(yes/no) > n
Run ICA-AROMA?
(yes/no) > n
Run postprocessing?
(yes/no) > y
Which postprocessing streams should be run? Press ENTER to select all.
1: postproc1
2: rest
Enter one or more numbers separated by spaces and then ENTER, or 0 to cancel
1: 1You can achieve the same thing by passing in arguments for
steps, postprocess_streams, and
subject_filter, which will schedule the jobs without
prompting you.
run_project(scfg, steps="postprocess", postprocess_streams="postproc1", subject_filter="540311")Prerequisites and Setup
BrainGnomes is designed to support end-to-end processing of fMRI data, from original DICOM files to postprocessed data ready for analysis. That said, you are not required to setup or run any given step. For example, if you have data that have already be processed up through fmriprep, you could use BrainGnomes only for postprocessing. In the setup phase, you will be asked for Singularity containers for all analysis steps, but many of these can be skipped if you have no intention of running that step (e.g., MRIQC).
Before using BrainGnomes, attend to the following
prerequisites:
- Installation: Install the BrainGnomes R package. The package is not on CRAN (yet), so for now, you must install from GitHub. Then load it in your R session:
# Install (if needed) and load BrainGnomes
# devtools::install_github("UNCDEPENdLab/BrainGnomes")
library(BrainGnomes)- Working Python installation: During postprocessing, the pipeline supports masking the data by a brain mask that matches the stereotaxic space. For example, if your fmriprep data are in MNI152NLin2009cAsym with a resolution of 2mm, a custom brain mask released with the template can be applied to the data to remove non-brain voxels. This relies on TemplateFlow (http://templateflow.org/), a Python library that provides a number of templates. For this step to work, you must have a working Python installation.
- HPC Access: Access to an HPC cluster with a job scheduler supported by BrainGnomes (currently SLURM or TORQUE). You should know which scheduler your cluster uses.
- Singularity: Singularity must be available on the HPC system
- You must have downloaded or built the required container image files for:
- fMRIPrep (e.g., a .sif image of fMRIPrep)
- HeuDiConv (container for DICOM to BIDS conversion)
- MRIQC (container for MRI quality metrics)
- ICA-AROMA (if you plan to use ICA-AROMA for denoising)
- BIDS Validator (either a container or an installed binary for the BIDS validation tool) Make note of the filesystem paths to each of these container files or executables, as the configuration will require them.
- Data and Files:
- DICOM files for your study, organized in subject (and optionally session) folders.
- A heuristic file for HeuDiConv (a Python script defining how to translate DICOM filenames into BIDS format). You or your lab should have this .py file prepared for your study’s naming conventions.
- A FreeSurfer license file (e.g., license.txt or FreeSurferLicense.txt). fMRIPrep requires a valid FreeSurfer license to run. You can obtain one for free from the FreeSurfer website. Save the license file path for the configuration.
- Optionally, a TemplateFlow directory if you want to use a non-default location for TemplateFlow data (standard brain templates). If unsure, you can specify an empty or new directory and fMRIPrep will manage downloading templates there.
With these in place, we can proceed to create a project configuration.
Creating a Project Configuration with
setup_project()
The first step in using BrainGnomes is to create a
project configuration, which is essentially a structured list containing
all the parameters and paths needed for your pipeline. This
configuration can be created interactively using the
setup_project() function. In typical use, you will call
setup_project() with no arguments to start a new
configuration from scratch, and it will interactively prompt you for all
required information. The function returns an object with your settings,
and it saves a a YAML configuration file to the root of the project
directory ($metadata$project_directory) called
"project_config.yaml". If you later
Let’s run setup_project() to initialize a new project
config:
# Start an interactive setup for a new fMRI project
scfg <- setup_project()This function function will ask a series of questions in the R console to gather information about your project. Below we outline the typical prompts and how to respond:
- Project Name – A short name for your project (used for labeling and logging). Example: “MyStudy2025”.
- Project Directory – The root directory where all project outputs will be stored. You can provide a path (absolute or relative). If the directory does not exist, BrainGnomes will offer to create it for you. For example: “/proj/Longleaf/MyStudy2025” (this will contain subfolders for BIDS data, fMRIPrep outputs, logs, etc.).
- DICOM Directory – The location of your raw DICOM files. This could be a folder containing subfolders per subject (and session). If it doesn’t exist, you’ll be prompted to create it as well. For example: “/proj/Longleaf/MyStudy2025/data_DICOMs”.
- TemplateFlow Directory – Path to your TemplateFlow data (standard templates for fMRIPrep). If you have a central TemplateFlow directory (e.g., ~/templateflow or a shared path), provide it.
- Scratch Directory – A path for temporary scratch space. This is often on a fast storage (like $TMPDIR or a scratch disk) that is not intended for long-term storage. If you’re not sure, you can use a sub-directory in your project directory (e.g., “/proj/Longleaf/MyStudy2025/scratch”). Just make sure to keep an eye on the size of this folder and clean it up periodically if this isn’t done automatically by your HPC system.
You will then be asked whether you want to include major steps in the pipeline. Each of these can be toggled depending on your intended use. For example, if you already have fmriprep-processed data and only wish to apply postprocessing, you wouldn’t need BIDS conversion. For each major step in the pipeline, you will be asked to specify the following HPC scheduler settings:
- The amount of memory needed for the job in GB.
- The number of hours needed to complete the job. (Always start a bit higher than you think you need to avoid killed jobs.)
- How many CPU cores should be requested for this job.
- Any other command line arguments that will be passed to the command (e.g., fmriprep)
- Any arguments that should be passed to the HPC scheduler (e.g., SLURM) beyond what the pipeline already handles itself.
Flywheel sync setup
If your DICOM data live in a Flywheel project, BrainGnomes can
optionally run a Flywheel-to-filesystem synchronization step before any
downstream processing. This uses the Flywheel CLI (fw sync)
to download DICOMs to your project so that BIDS conversion (and later
steps) operate on a complete, current dataset.
- Enable in setup: Answer “Run Flywheel sync?” with yes in
setup_project()(or useedit_project()→ “Flywheel Sync”). You will be prompted for:- Flywheel project URL (
source_url), for example:fw://hallquistm/momentum/. Watch out for the trailing slash! Flywheel sync operates a lot like rsync, so having the trailing slash here means, “synchronize files within momentum.” - Drop-off directory for downloaded DICOMs
(
metadata/flywheel_sync_directory). This is typically your project DICOM folder. - Temporary directory for transfers
(
metadata/flywheel_temp_directory). Defaults under your scratch directory. - Whether to save audit logs
(
flywheel_sync/save_audit_logs). When enabled, a CSV is written under your projectlogs/. - Location of the Flywheel CLI binary
(
compute_environment/flywheel), i.e., the path to thefwexecutable.
- Flywheel project URL (
- Get an API token: The Flywheel CLI requires an API key associated
with your user. Generate one in the Flywheel web UI:
- Navigate to your Flywheel site (for example,
https://flywheel.example.edu). - Open your user profile → API Keys and create a key. A direct route
in many deployments is
https://<your-flywheel-host>/#/profilethen the API Keys tab. - Copy the generated token and keep it secure.
- Navigate to your Flywheel site (for example,
- Log in with the Flywheel CLI before syncing: BrainGnomes does not
perform
fw loginfor you. You must complete CLI login once on the machine/account that will run the jobs, e.g.:
fw login <YOUR_API_TOKEN>After successful login, the CLI stores credentials locally so
subsequent runs can use fw sync non-interactively. If
fw login is not complete, the Flywheel sync step will
fail.
What BrainGnomes runs: The sync job uses fw sync with
sensible defaults (includes DICOMs, non-interactive -y,
your specified temp path, and optional audit log). Data are written to
your flywheel_sync_directory. Typically the call will look
something like this:
/path/to/fw sync --include dicom -y --tmp_path <scfg$metadata$flywheel_temp_directory> \
--save-audit-logs <logs/flywheel_sync_audit.csv> \
fw://hallquistm/momentum/ \
/proj/mnhallqlab/studies/momentum/data/fMRI_MRI/raw_images/unc_flywheel_syncA note about trailing slashes in the fw:// string
Flywheel sync operates a lot like rsync, so having the trailing slash
in the fw:// project string like
fw://hallquistm/momentum/ indicates that folders within
that project are synchronized to the top level of the
flywheel_sync_directory (in the example,
/proj/mnhallqlab/studies/momentum/data/fMRI_MRI/raw_images/unc_flywheel_sync).
Thus, you typically want a trailing slash after your project name in the
fw:// string so that the top-level folder itself isn’t
copied to the synch folder.
Deferred queueing of subject processing when flywheel comes first
Usage tip: You can include Flywheel sync as part of a larger unattended run, for example:
run_project(scfg, steps = c("flywheel_sync", "bids_conversion", "fmriprep"))When flywheel_sync is in the requested steps,
BrainGnomes defers subject-level job scheduling until after the sync job
completes. This ensures downstream steps (BIDS conversion, fMRIPrep,
etc.) see all current data from Flywheel.
BIDS conversion
You will be prompted for whether you want to include BIDS conversion:
Run BIDS conversion? (yes/no; Press enter to accept default: yes).
If you say yes, you will be asked for the location of a heudiconv
container, followed by questions about memory, compute hours, CPU cores,
command line arguments, and scheduler arguments.
BrainGnomes uses HeuDiConv (https://github.com/nipy/heudiconv) to convert DICOM
images into BIDS-compatible files and folders. HeuDiConv requires
information on how to identify subjects/sessions in your DICOM folder
and how to convert them. As part of the BIDS conversion setup, you will
be asked to provide:
Subject Regex: A regular expression used to match all subject directories in the root of your DICOM directory. Consider, for example, a DICOM directory that includes the following folders:
logs,5300, and5420, where the numbered directories are the subject IDs. In this case, a good regular expression for identifying subjects would be,[0-9]+– matching folders containing numbers. If you want any folder in the root of your DICOM directory to be considered a subject folder, use.*.(Optional) Session Regex: if you have multi-session data and these are stored in your DICOM directory as subfolders (i.e., something like sub-100/ses-1, sub-100/ses-2) you may specify a regular expression to match session folders inside each subject’s folder. For example, a session regex of
ses-[0-9]+would match any subfolder starting withses-followed by one or more numbers. If you do not have multisession data in this form, leave this blank.Subject ID Match Regex: Whereas the Subject Regex specifies what folders in the root of your DICOM directory should be considered, the Subject ID Match regular expression specifies how to extract the ID from the folder name. Use parentheses to denote which part of the folder name should be preserved as part of the ID. For example, if folders are named something like
sub-<numbers>and you want to keep the numeric part as the ID, the Subject ID Match Regex would be,sub-([0-9]+). If the ID is stored in different parts of the folder name, you can use more than one set of parentheses, in which case these will be extracted and pasted together with an underscore separating each part. For example, consider a foldersubject-16-visit-3. You could extract a subject ID of16_3using the Regex:subject-([0-9]+)-visit-([0-9]+).Session Match Regex: This follows the same logic as the Subject ID Match Regex, but applies to session folders nested within subject folders. For example, if you have a folder like:
subject-12/ses-1, you would specifysubject-([0-9]+)as your subject ID match regex andses-([0-9]+)as your session match regex.Heuristic File: Path to the heudiconv heuristic Python script for your project. For example:
"/proj/Longleaf/MyStudy2025/heuristic.py". This script defines the naming scheme for converting DICOMs to BIDS format. Details for preparing such files can be found here: https://heudiconv.readthedocs.io/en/latest/heuristics.html.Overwrite and Clear Cache: Boolean flags (overwrite and clear_cache) that determine if heudiconv should overwrite existing converted data and clear any caching between runs. Usually, you can accept defaults (FALSE) unless you are re-running conversion and want to start fresh.
fmriprep setup
Next, you will be asked to decide whether to include fmriprep in the
pipeline:
Do you want to include fMRIPrep as part of your preprocessing pipeline? (yes/no; Press enter to accept default: yes).
If you say yes, you will be asked for the following:
- Location of the fmriprep container
- Resource requirements: Defaults for BrainGnomes fmriprep are 48 GB RAM, 24 hours, and 12 cores
- Additional command line arguments to fmriprep. See https://fmriprep.org/en/stable/usage.html for details.
- Additional arguments to be passed to the HPC scheduler
- Output Spaces: You will choose the standard spaces that fMRIPrep should output the preprocessed data in. Common choices include MNI anatomical spaces (e.g., MNI152NLin6Asym), the subject’s T1w native space, fsaverage (for surface data), etc. The setup_project() function will present a menu or prompt for selecting these. You can pick multiple spaces. For example, a typical selection might be MNI152NLin6Asym and T1w (and if analyzing surface data, possibly fsaverage).
- You will also be asked to provide a ‘resolution index’ for each template, which governs the spatial resolution of the output image. If this is omitted, the resolution of the output file will match the voxel size of the input file. Note that the index is not synonymous with the voxel size (e.g. ‘2’ may not be 2mm). See https://fmriprep.org/en/stable/spaces.html#standard-spaces for details.
- FreeSurfer License File: You must provide the path to your FreeSurfer license file so fMRIPrep can run FreeSurfer during preprocessing.
MRIQC setup
Next, you will decide whether to include MRIQC in the pipeline:
Run MRIQC? (yes/no; Press enter to accept default: yes). If
you say yes, you will be asked for:
- Location of the mriqc container
- Resource requirements: Defaults for BrainGnomes mriqc are 32 GB RAM, 12 hours, and 1 core
- Additional command line arguments to mriqc. See https://mriqc.readthedocs.io/en/latest/usage.html#command-line-interface for details.
- Additional arguments to be passed to the HPC scheduler
ICA-AROMA setup
Next, you will decide whether to include ICA-AROMA in the pipeline:
Run ICA-AROMA? (yes/no; Press enter to accept default: yes).
If you say yes, you will be asked for:
- Location of the ICA-AROMA container
- Resource requirements: Defaults for BrainGnomes mriqc are 32 GB RAM, 36 hours, and 1 core
- Additional command line arguments to fmripost_aroma. See https://fmripost-aroma.readthedocs.io/latest/usage.html#command-line-arguments for details.
- Additional arguments to be passed to the HPC scheduler
(Note: As of fMRIPrep v20.2+, ICA-AROMA is no longer integrated in fMRIPrep and must be run separately as a BIDS-App called fmripost-aroma.)
Postprocessing setup
Next, will decide whether to include postprocessing in the pipeline:
Do you want to enable postprocessing of the BOLD data?.
Postprocessing includes several optional steps, including
- Applying a brain mask
- Spatial smoothing
- Denoising using ICA-AROMA
- ‘Scrubbing’ of high-motion/high-artifact timepoints
- Temporal filtering (e.g., high-pass filtering)
- Intensity normalization
- Confound calculation and regression
If you say ‘yes’ to postprocessing, you will be asked to setup one or more postprocessing streams for your data. BrainGnomes supports multiple postprocessing streams, such that the same data can be postprocessed in multiple ways. For example, you might wish to compare results with different levels of smoothing or with different temporal filtering settings.
To specify streams, you will be dropped into a menu system that lookes like this:
Postprocessing supports multiple streams, allowing you to postprocess data in multiple ways.
Each stream also asks about which files should be postprocessed using the stream. For example,
files with 'rest' in their name could be postprocessed in one way and files with 'nback' could
be processed a different way.
Current postprocessing streams:
(none defined yet)
Modify postprocessing streams:
1: Add a stream
2: Edit a stream
3: Delete a stream
4: Show stream settings
5: FinishFor additional details about postprocessing, see (in progress) Postprocessing vignette.
ROI time series extraction and functional connectivity calculation
Finally, you will be asked whether to include ROI extraction and functional connectivity calculation in the pipeline. This step allows you extract average time series from regions of interest (ROIs) in one or more atlases/ROI masks. You can also ask BrainGnomes to compute correlations among the ROI timeseries for functional connectivity analyses.
The extract_rois() function – called using
run_project – loops over configured atlases and writes out
ROI time series and correlation matrices to the project’s
data_rois directory using BIDS-style filenames. For more
details about ROI reduction choices and correlation options, see the Extracting ROIs vignette.
BIDS validation setup
As an optional part of the pipeline, bids-validator can be run on the
BIDS directory (once BIDS conversion has completed). This is
accomplished by run_bids_validation(), which can be run as
needed. Here, saying yes to the prompt
Enable BIDS validation? (yes/no; Press enter to accept default: yes)
will setup the bids-validator step for later use. You will be asked
for:
- Location of the bids-validator program
- Resource requirements: Defaults for BrainGnomes bids-validator are 32 GB RAM, 2 hours, and 1 core.
- Additional command lines arguments to bids-validator. https://bids-validator.readthedocs.io/en/stable/user_guide/command-line.html for details.
- Additional arguments to be passed to the HPC scheduler
Conclusion
After setup_project completes, it will write a file
called project_config.yaml into your project directory (if
one exists, you will be prompted whether to overwrite it). This will
contain all of your settings and can be used in future to load a project
into R from an existing configuration.
BrainGnomes workflow in a nutshell
Step 1: Setup your project
Use setup_project() to interactively complete or correct
configuration entries:
scfg <- setup_project()This will say a project_config.yaml file in your project
directory.
Step 2: Load your configuration
The step above results in an object in your R workspace called
scfg that contains all of your project settings. This is
great for now, but if you quit R and come back later, you probably want
to pick up from where you left off. You can use
load_project() to load an existing configuration:
scfg <- load_project("/path/to/my/project")Step 3: Edit configuration
Use edit_project() for a menu-driven interface to modify
parts of the project configuration.
scfg <- edit_project(scfg)Follow the prompts to choose what to edit. This is useful if you want to change job resources (e.g., memory or CPU for fMRIPrep) or adjust CLI options for specific pipeline steps.
Step 4: Run the project
Once your configuration is complete, you can run the pipeline.
run_project(scfg)By default, this will prompt you whether to run all steps in your pipeline. It will also process all available subjects and sessions defined in the BIDS directory.
If you want to run only a subset of subjects, you can use the
subject_filter argument like:
# run subjects 242 and 510 only
run_project(scfg, subject_filter=c("242", "510"))Likewise, if you want to skip the menu prompts for which step to run,
you can use the steps argument. For example, to run only
postprocessing on these subjects:
# run subjects 242 and 510 only
run_project(scfg, subject_filter=c("242", "510"), steps="postprocess")The available steps are
"bids_conversion", "mriqc", "fmriprep", "aroma", "postprocess", "extract_rois".
Note that if you request a step in run_project that was
never configured it will result in an error.
Running BrainGnomes on the command line
As of August 2025, this is a work in progress. That said, there is basic support for using BrainGnomes on the Linux command line. This is helpful if you prefer not to start an R session or you just want to perform basic operations such as editing a study configuration or running a processing step.
To get BrainGnomes on the command line, you need to add the location of the package to your Linux path. If you don’t know where it is installed, run this in an R session.
find.package("BrainGnomes")You then need this in your path when a terminal starts. If you use
bash, the easiest route is often to add a line like this to your
~/.bashrc.
export PATH="/Library/Frameworks/R.framework/Versions/4.5-arm64/Resources/library/BrainGnomes:$PATH"This will enable you to type BrainGnomes at the terminal
prompt:
> /BrainGnomes
Usage: BrainGnomes <command> [options]
Commands:
setup_project [project_name] [project_directory]
edit_project <project_directory|config.yaml>
run_project <project_directory|config.yaml> -steps <steps> -subject_filter <ids> -postprocess_streams <streams> [-debug] [-force]As seen above, the CLI supports setup_project,
edit_project, and run_project. The basics of
these commands have already been described above. On the CLI, the only
difference is that objects such as scfg are not persisted
in a session. Rather, changes are made to the
project_config.yaml script in the project directory.